Де не справляється людська – впорається бактеріальна імунна система (початок)
22 Грудня 2014 | Молекулярно про Рак
Категорія: Медицина
Теги: CRISPR, ex vivo, імунна система, ВІЛ, генна терапія, геномне редагування, снід, стовбурові клітини
Вірус імунодефіциту людини (ВІЛ) атакує клітини імунної системи, а саме CD4+ Т-хелпери, залишаючи організм людини на поталу збудникам найрізноманітніших інфекцій. Проте, наскільки б неймовірним не здавалося б вам таке твердження, на допомогу людині в найбільш скрутну годину приходить імунна система бактерії. Згаданий вірус оповитий туманною аурою різноманітних міфів: ніби-то його випустили ЦРУ для контролю кількості населення Африки, чи це зробили китайці для з тією самою метою проти США, а інтернет-повідомлення про винайдення чудодійних ліків проти ВІЛ з’являються чи не щогодини. У той же час, щороку в світі публікуються тисячі наукових робіт із дослідженнями наноскопічного вбивці. Але вірус із холоднокровною байдужістю ставиться до тієї шани, котру віддає йому вищий примат виду Homo sapiens sapiens, і тишком-нишком робить свою вбивчу справу. Ми ж з вами залишимо осторонь спекуляції та змови, які, звичайно, не мають нічого спільного із реальністю, і критично оцінимо можливості застосування бактеріальної системи захисту проти фагів CRISPR/Cas9 в боротьбі із ВІЛ/СНІДом, мимохідь смакуючи найкрасивіші моменти з історії генної терапії цієї недуги.
В середині 90-х років минулого сторіччя стало відомо, що ВІЛ використовує рецептор CD4 та корецептор CCR5 для входження в клітину. Цим, власне, зумовлена його специфічність до Т-хелперних клітин та деяких дендритних клітин. ВІЛ є ретровірусом, це означає, що він існує як в цитоплазмі клітини у вигляді РНК, так і в стані інтегрованому в геном клітини, тож інфекція протягом тривалого часу протікає латентно. Проте в якийсь момент віруси масово переходять від лізогенної до літичної фази розвитку, що має наслідком швидку загибель клітин-носіїв, власне, CD4+ Т-клітин та дендритних клітин. Оскільки останні виконують інтегральну роль в розвитку імунних реакцій людини, то виведення їх з ладу веде за собою розвиток імунодефіциту. На рисунку нижче підсумовано шляхи передачі ВІЛ від клітини до клітини.
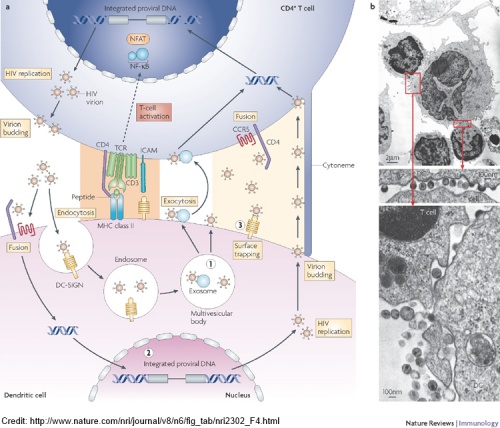
CCR5 (Cysteine-cysteine chemokine receptor 5) є рецептором так званих хемокінів – молекул, які беруть участь в підтриманні запалення та спрямовуванні клітин до «гарячої точки імунного протистояння». Протягом останнього часу саме він привернув увагу вчених як можлива ціль генної терапії ВІЛ/СНІДу, і на це були свої причини. По-перше, CCR5 є абсолютно необхідним для входження ВІЛ всередину клітини, що підтверджується кількома дослідженнями, основні з яких приведені нижче. По-друге, його роль в регуляції імунітету, як видається, не є суттєвою – його функції дубльовані іншими рецепторами хемокінів. На відміну від більшості генів, втрата експресії CCR5 не викликає суттєвих порушень. Отже, можна без особливого ризику для організму вимкнути ген, що кодує протеїн CCR5, і таким чином позбавити вірус імунодефіциту можливості передаватися від клітини до клітини.
Приклад 1. Резистентність до вірусу носіїв гомозиготної мутації CCR5-Δ32, яка є делецією 32-х пар основ з 794 по 825 нуклеотид. Така делеція по суті є нонсенс мутацією, призводячи до зсуву рамки зчитування і появи передчасного стоп-коду. Відповідно, мРНК рецептору не транслюється, а піддається нонсенс-опосередкованому розпаду (nonsense-mediated decay). Тож, такі індивіди не експресують CCR5 на поверхні імунних клітин, почуваються добре і не хворіють на СНІД. За даними генетиків носіями такої мутації є до 18 % жителів Європи, а в її північній частині до 20 %. Існує припущення, що висока частота такої мутації спричинена селективним тиском на людську популяцію таких патогенів як Yersinia pestis (збудник чуми) та Variola virus (збудник віспи) протягом тривалого часу.
Приклад 2. У 2008 році пацієнту із ВІЛ/СНІД, в якого на фоні імунодефіциту спостерігався розвиток гострої лімфобластної лейкемії, було здійснено трансплантацію кісткового мозку від здорового донора, гомозиготного за CCR5-Δ32 мутацією. Операція пройшла успішно, і заразом із лейкемією його таким чином вилікували від ВІЛ. Точніше сказати, у нього спостерігався різкий спад кількості вірусу в крові, але, як відомо, пацієнти після трансплантації від неродинного донора повинні постійно приймати імуносупресанти для попередження можливого відторгнення. Тож, така цинічна іронія долі.
Приклад 3. Під час дослідження хворих на гемофілію, які отримували фактор VIII із сироватки крові людини, виявилося, що вони були несприйнятливі до ВІЛ, який також випадково містився в цій сироватці. Дослідження проводилося в середині 80-х, коли про ВІЛ лишень дізналися – обов’язкового скринінгу донорів сироватки тоді ще не проводили. З’ясувалося, що у пацієнтів-гемофіліків були підвищені рівні експресії бета-хемокінів. Останні конкурували із лентівірусом (у тому числі ВІЛ) за зв’язування з CCR5 рецептором, не даючи йому шансу пробратися в клітину.
Станом на 2007 рік Національний Інститут Здоров’я Сполучених Штатів сумарно профінансував три десятки лабораторій на суму 10 мільйонів докризових доларів, поставивши їм завдання розробити ефективну генну терапію від СНІДу. Це не враховуючи 14 мільйонів, які отримав нобелівський лауреат Девід Балтімор на розробку супер-антитіла проти вірусу імонудефіциту. Тож, як бачимо, американські вчені достатньо щільно взялися за цю проблему.
Загалом усі підходи генної терапії СНІДу мають кілька схожих етапів: вчені забирають клітин крові пацієнта з коровотоку або безпосередньо з кісткового мозку, збагачують препарат на клітини, на які саме націлений вірус, вносять генетичні модифікації в них різними шляхами, та повертають клітини назад пацієнту. Головні відмінності полягають в типі генного вектора, що використовується для здійснення модифікації, а також в способі культивування клітини перед процедурою трансфекції, що в свою чергу залежить від ступеню диференційованості клітин, які беруться – попередники мієло- та лімфопоезу, які вже стали на шлях диференціювання (так звані «committed progenitor cells»), або ж недиференційовані стовбурові клітини кровотворення. Ключем до успішності терапії є як ефективна трансформація великої кількості клітин, так і виживаність клітин після повернення до організму пацієнта. Слід зазначити, що існують численні стратегії генної терапії ВІЛ, але, за неможливістю охопити їх усі, ми зосередимось саме на тих, що спрямовані проти гену CCR5.
Коротко про основні здобутки на цій ниві станом на почак 2000-х. Steinberger et al. (2000) розробив внутрішньоклітинне одноланцюгове антитіло (так зване intrabody), яке зв’язує протеїн CCR5 під час його транспорту по секреторній системі клітини, таким чином затримуючи його і не даючи досягнути поверхні клітини. В теорії це мало б викликати набуття клітиною CCR5–/– фенотипу навіть без втручання в структуру самого гену. Strizki et al. (2001) синтезували малу молекулу, що блокує CCR5-опосередковане входження вірусу в клітину. Це зовсім не генна терапія, але вона також спрямована проти згаданого хемокінового рецептору. Препарат продемонстрував 50-60 відсоткову біодоступність у разі орального застосування і час напіврозпаду в сироватці до шести годин на тваринних моделях. Також були повідомлення (Qin et al., 2003) про застосування малих шпилькових РНК (siRNA) проти мРНК CCR5. Був розроблений лентівірусний вектор для доставки послідовності siRNA до імунних клітин. За іронією долі використовується той самий вірус СНІДу тільки в зміненій формі.
Але усі перелічені стратегії не виявилися достатньо ефективними (в іншому разі, з огляду на роки публікацій, ми б вже давно знали про існування ліків від СНІДу). Фармакологічне подавлення рецептору потребує постійного прийому достатньо великих доз препарату, додатково до того «коктейлю», який зазвичай приймається хворим на імунодефіцит. До того ж у підсумку виникає резистентність вірусу. Підходи з intrabody та siRNA передбачають процедуру вставляння нових генів у геном клітин крові людини, що почасти пов’язано із двома серйозними проблемами – низькою ефективністю модифікації та ризиком для життя пацієнта. По-перше, принципово неможливо досягнути 100% трансформації усіх клітин (Т-хелперів та дендритних клітин). Насправді, вірогідність вставки гену навіть шляхом гомологічної рекомбінації настільки низька, що просто не дозволить отримати потрібну кількість модифікованих клітин, де intrabody або siRNA експресувалися б достатньо інтенсивно для успішного пригнічення CCR5. Але це, ще півбіди, оскільки навіть така непевна вставка може втрачатися (через ще одну рекомбінаційну подію або епігенетичний сайленсінг) протягом першого ж року після терапії. І в такому випадку синтез функціонального CCR5 рецептору буде поновлюватися. По-друге, виникає небезпека інсерційного мутагенезу. Наприклад, у 2005 році з’явилося повідомлення про розвиток лейкемії у дітей, що отримували генну терапію від СНІДу (як вектор використовувалися мишачі ретровіруси). Лентівірусні вектори працюють краще і вважаються безпечнішими за ретровіруси лейкемії мишей, проте у разі використання ВІЛ-подібних вірусів для доставки генетичних конструкцій виникає ще одна страшна небезпека: між хвороботворним та терапевтичним вірусами може відбутися рекомбінація, наслідки якої складно передбачити. На рисунку нижче, узагальнено усі наявні наразі стратегії пригнічення експресії рецептору CCR5.
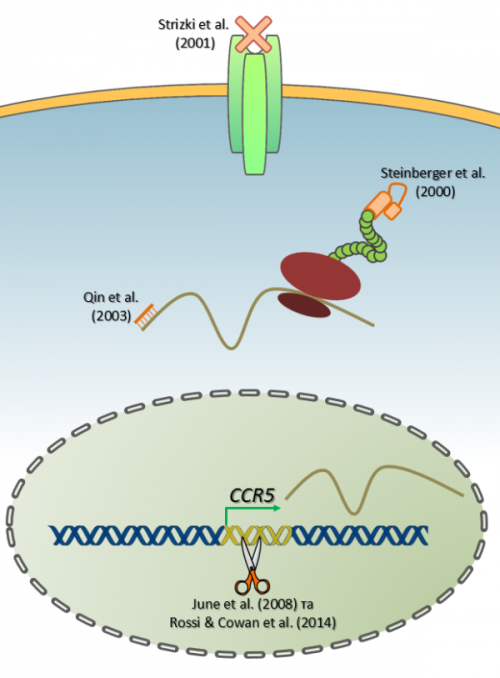
У 2005 на обрії з’явилася технологія, покликана подолати усі вищезазначені проблеми, її назва – геномне редагування. Винайдення модульних едонуклеаз із програмованою специфічністю, таких як нуклеази із ДНК-зв’язуючими доменами «цинкові пальці» (zinc-finger nucleases, ZFN) та трансактиватор-подібні ефекторні нуклеази (Transcription activator-like effector nucleases, TALENs), надало можливість напряму маніпулювати із генами безпосередньо на клітинній ДНК. За принципом «зламати легше, ніж збудувати», такий підхід значно спрощує нокаутування генів – достатньо лише нетривалої експресії ZFN в клітині для того, щоб незворотньо пошкодити певний клітинний ген. Тож, не дивно, що цією стратегією вирішили скористатися борці зі СНІДом. Взагалі, редагування геному, ZFN та TALENs достойні окремого огляду на сторінках Моєї Науки {може іншим разом ;)}.
Перша спроба геномного редагування за допомогою ZFN нуклеаз у Т-клітинах була здійснена June et al. (2008). Тимчасова експресія в складі аденовірусного вектора ZFN протеїну, націленого на ген CCR5, викликала ефективне і стабільне нокаутування цього гену, що спадкувалося в ході подальшої проліферації цих клітин. Цікавою знахідкою було те, що нокаут CCR5 надає селективну перевагу CD4+ T-клітинам у разі їх культивування із вірусом імунодефіциту in vitro. Надалі вчені спостерігали схожі результати in vivo, використовуючи мишачу модель ВІЛ. Вони продемонстрували, що пересадження ВІЛ-інфікованим мишам ZFN-модифікованих CD4+ T-клітин призводить до значного зменшення вірусного навантаження на фоні зростання кількості CCR5–/– Т-клітин в довгостроковій перспективі. Не гаючи часу, дослідники взялися до реалізації цієї стратегії в клініці. В період з травня 2009 по липень 2012 було залучено 12 ВІЛ/СНІД-пацієнтів, а результати дослідження були опубліковані нещодавно (березень 2014) в журналі New England Journal of Medicine. Подібно до попереднього експерименту, спочатку в пацієнтів було зібрано CD4+ T-лімфоцити, потім в них було виконано нокаут CCR5 гену шляхом тимчасової експресії специфічної ZFN нуклеази із подальшим поверненням модифікованих клітин назад до кровотоку пацієнтів. За 4 тижні після вдалого перенесення клітин 6 з 12-ти пацієнтів були зняті із супроводжуючої антиретровірусної терапії з метою дослідження динаміки CD4+ Т-хелперних клітин. Виявилося, що за 12 тижнів без супутньої терапії кількість Т-хелперів дикого типу різко скоротилася на фоні незначущих коливань кількості модифікованих Т-хелперів, що знову свідчило про позитивну селекцію щодо CCR5–/–. Протягом 254 днів випробувань показники вірусного навантаження в крові пацієнтів впали на три порядки (в 1000 разів), але повного очищення від вірусу імунодефіциту вдалося досягти лише у одного пацієнта. Як виявилося пізніше, він був гетерозиготним за CCR5-Δ32 мутацією, тож ефективність подвійного нокауту (CCR5–/–) в Т-клітинах цього індивіда була вдвічі вищою, ніж у інших. Це зайвий раз підтверджує важливість згаданого корецептора як специфічної цілі для генної терапії ВІЛ/СНІДу.
Це була, так би мовити, передісторія, а тепер — власне, до CRISPR/Cas9.
Дні науки

Наші проєкти

Щеплення Правдою



Обговорення